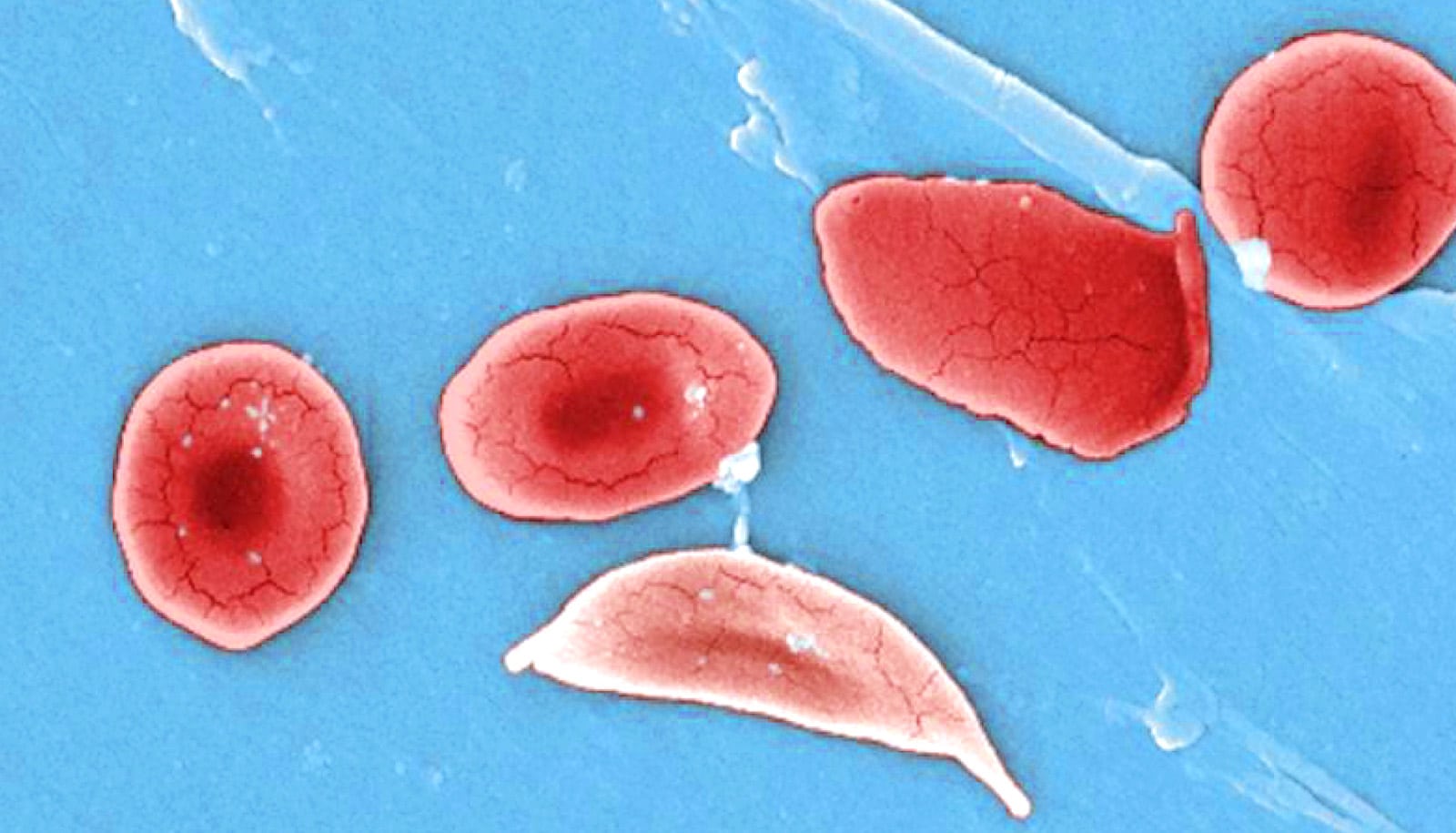
A new study suggests using CRISPR-Cas9 and a corrective short DNA template could offer a safe and efficient way to to repair the mutation that causes sickle cell disease.
Sickle cell disease, which affects about 100,000 Americans and millions worldwide, is a painful and often fatal inherited condition. A single mutation in hemoglobin subunit beta (aka beta-globin) forces normal, disc-shaped red blood cells to stiffen and take characteristic “sickle” shapes. These cells can damage vessel walls and clot small blood vessels, stopping the delivery of oxygen to tissues.
Today, some people with the disease receive stem cells from a matched, related donor, but that’s an option available to fewer than 15 percent of patients, researchers say. Modifying the patient’s own hematopoietic stem and progenitor cells (HSPCs) is a better strategy—an option theoretically available to every patient because the risk of rejection wouldn’t exist.
Ideally, stem cells isolated from the patient’s bone marrow would be gene-edited and tested, and chemotherapy would reduce the patient’s stem cells to make room for the edited cells. The corrected cells would then go back into the patient, where they could proliferate and spawn healthy blood cells.
Gang Bao, professor of bioengineering at Rice University’s Brown School of Engineering, tested patient HSPCs in rodents and demonstrated that a fraction of the gene-edited cells from patients could survive and function for about 4 months.
Vivien Sheehan, an assistant professor of pediatrics-hematology/oncology and hematology at Baylor College and a member of the sickle cell program at Texas Children’s Hematology Center, obtained the stem cells from the peripheral blood of five patients and the bone marrow of two patients with the disease.
Her lab characterized the type of hemoglobin the edited cells made and showed that gene editing can provide enough protective and normal hemoglobin to prevent sickling, even under the severe hypoxia that promotes it.
“The good news is that normal red blood cells have a lifespan about nine times that of the sickle cells.”
Early tests using wild-type Cas9 protein—the “scissors” that target and cut specific sections of DNA—from Streptococcus pyogenes led to stem cells with high levels of unintended DNA edits. These off-target edits included large chromosomal deletions and inversions that could cause disease.
In a second round of tests, researchers adopted a more recent “high-fidelity” version of Cas9, which Integrated DNA Technologies developed. That led to significantly reduced off-target edits, Bao says.
Gene editing doesn’t fix all the stem cells isolated from a patient with the sickle mutation, researchers say.
At best, according to experiments in the Bao lab, it fixed up to 40 percent of them. In another 50 percent of cells, DNA was cut and not repaired by the corrective DNA template, but that appeared to boost the stem cells’ expression of fetal hemoglobin (HbF), a type a different gene that does not carry the sickle mutation and is normally turned off a few months after birth.
Researchers know that fetal hemoglobin mutes the effects of sickle cell disease. Bao suspects that if the expression of fetal hemoglobin in edited cells persists, 90 percent of the stem cells from patients will either have the sickle mutation fixed, or have sickling prevented by fetal hemoglobin.
“Our hope is that if we take out a fraction of HSPCs from a patient’s bone marrow and damage the remaining ones, then edit the HSPCs and deliver them back, the combination of gene-corrected and HbF-expressing cells will be enough to cure the disease,” Bao says.
That 10 percent of stem cells continued to produce sickle cells should not be a big concern, he says. “The good news is that normal red blood cells have a lifespan about nine times that of the sickle cells,” Bao says. “That means over time, the majority of the red blood cells would be normal cells. That’s the scenario we would like to see.”
The researchers don’t yet know if cutting beta-globin to boost fetal hemoglobin levels would provide a long-lasting benefit or whether it risks inducing beta thalassemia, a blood disorder that reduces the production of hemoglobin. “There is a risk, and we need to understand it better,” Bao says.
It is also unclear how many stem cells need editing to provide long-term relief to a patient, Bao says.
“We don’t know what percentage of the HSCs with gene correction could effectively treat sickle cell disease. Some people say just a few percent, and some say 5-10 percent. My guess is around 5 percent would be sufficient, but the exact percentage could only be established through clinical trials.”
The road to human trials may still take some time, Bao says.
“Technology-wise, we are ready to do a clinical trial,” he says. “But just getting approval for a trial will require a lot of resources. We need to compile the data and do additional experiments to address the safety issues.”
Rice graduate student So Hyun Park and research scientist Ciaran Lee are co-first authors of the paper, which appears in Nucleic Acids Research. Additional coauthors are from Rice, the Texas Children’s Hematology Center/Baylor, and Stanford University.
The Cancer Prevention and Research Institute of Texas, the National Heart, Lung and Blood Institute, and National Institute of Diabetes and Digestive and Kidney Diseases of the National Institutes of Health, the Chao Physician-Scientist Award, the Stanford Maternal and Child Health Research Institute, the Amon G. Carter Foundation, a Laurie Kraus Lacob Faculty Scholar Award in Pediatric Translational Research, and the Sutardja Foundation funded the work.
Source: Rice University

